


Dreampore - Nanopore Protein Sequencing
Company
Dreampore is a French Nanopore Protein sequencing company based in Paris.
There’s not much information available on Dreampore, most of it comes from a Genomeweb review from December 2019. In this article they state that Dreampore has raised €600,000, and they have four employees. As far as I can tell from their company registration they were founded in 2018 and currently have 3 to 5 employees. According to LinkedIn, the CEO (Luc Lenglet) is also leading two other companies. I could only see one current employee on LinkedIn who appeared to be fully dedicated to the company.
Technology
Surprisingly I wasn’t able to find a patent covering the work presented in their Nature paper on protein sequencing. So this review is based on the publication only.
The work uses a protein nanopore, and detects molecules as they pass through and block a bias current. This is much like other forms of nanopore DNA sequencing.
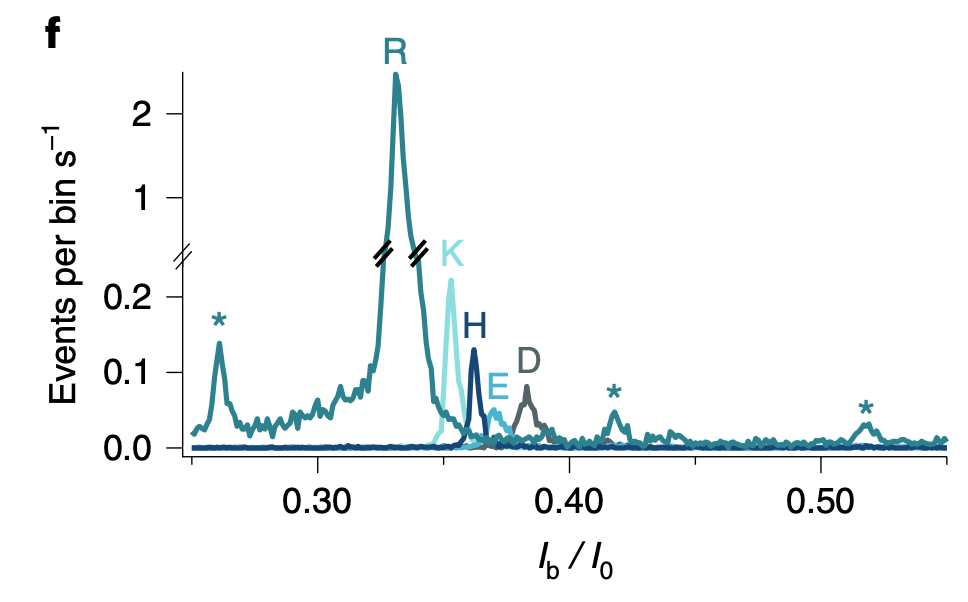
The publication builds on a previous work where they detect translocations of >7mer arginine (R) homopeptides. I’ll be covering this in a future post, because it’s kind of interesting in its own right. But essentially these RRRRRRR peptides block the aerolysin pore for a detectable duration. In the sequencing paper they use xRRRRRRR peptides where the x position varies. The poly-arginine region helps the peptide stick around in the pore long enough to be detected. But the idea is that the blockage current varies enough based on the single differing position.
And histograms suggest that in most cases it does:
When you look at the full set of amino acids, current blockages are less well separated:

The plots above use Ib/I0. This appears to be the signal normalized against the baseline current. It’s not super common to do this, and I wonder why have normalize against the baseline, rather than just measuring the offset against the baseline in pA. Possibly their measurements vary significantly with buffer concentration…
The raw ABF files (which suggests measurements were taken on a Axopatch) are available. So it’s possible to confirm this. But the scaling makes the plots a little harder to interpret. From example traces it looks like blockages are probably between 60 and 70pA (they all appear to be 0.3 and 0.4 in scaled units, and a typical baseline current appears to be ~100pA). So, you’re cramming 20 states into ~10pA. The best you’re like to do in terms of noise is likely ~1pA RMS at 10KHz.
They have a plot in the supplementary information which shows that in practice, they get about 10 pA of peak-to-peak noise on blockages.

From the supplementary information the average dwell time seems to be ~5ms (which remember is for 8 amino acids). So, let’s say 1ms per AA. So if we average down to 1KHz, we can probably get this to ~1pA of noise.
It seems likely that if they attempted sequencing, multiple positions are likely contributing to the signal. Let’s be conservative and say 3 positions. For 20 AAs that means 8000 possible combinations. So I’d speculate this comes down to:
0.001pA difference between each state and 1pA of noise
Which seems like a very hard problem to solve. Certainly one or two orders of magnitude harder than nanopore DNA sequencing.