


Twinstrand Biosciences
Business
Twinstrand is a University of Washington spinout based in Seattle. Crunchbase lists Twinstrand as being founded in 2015 which is not long after the foundational work was done (in 2012), they’ve recently raised a series B of $50M bringing their total to $73.2M. I see 62 employees on LinkedIn.
Approach and Applications
The basic play is that Illumina sequencing has an error rate that’s too high for some applications. To me, this is was kind of surprising. In Illumina sequencing, around 90% of bases are Q30. That’s an error rate of 1 in 1000. Do you really need an error rate lower than this? Twinstrand propose a number of applications, these are largely around very low level mutations.
Detecting residual acute myeloid leukemia (AML) after treatment.
Mutagenesis assays, for chemical and drug safety testing.
Cellular Immunotherapy Monitoring
In general, I’m used to seeing plays (like GRAIL) around cancer screening. But this is aimed more at cancer monitoring. The US national cost of cancer care is $150B, there are around 1.8M cancer cases. So, if we assume that this test will be required for cancer monitoring of every patient, and yields $1000 in profit that’s $1.8B in profit. Probably enough to support the company, and make investors happy…
But for the Twinstrand play to work, and justify a healthy valuation, at least the following needs to be true:
“ultra-high accuracy” is needed for cancer monitoring.
The Twinstrand approach is a practical method of generating “ultra-high accuracy” reads.
The Twinstrand approach is the only and best way to get “ultra-high accuracy”.
The first may be true, but it’s obviously not what GRAIL and other players have been working on for early stage cancer screening, where the focus has shifted toward base modification/methylation.
As to Twinstrand’s practicality? Hopefully we can gain some insight by reviewing the approach.
Technology
The technique relies on adding two pieces of information to double stranded DNA. The first is a unique index (a UMI) which uniquely identifies each double stranded fragment. The second is a strand-defining element (SDE). This a marker that allows the two strands forming a double stranded fragment to be distinguished.
Twinstrand use two UMIs. One of each end of the original double stranded fragment. They call these two UMIs “α” and “β” in the figure below.

The Y shaped adapters (labelled Arm 1,2) in the diagram above introduce an asymmetry between the strands. This provides the strand-defining element (SDE) described above.
To make this clearer I decided to break to the diagram further, showing the individual amplification steps involved:

Post amplification, and in 5’ orientation you will get 4 distinct read types as shown above. Each of these can be classified as coming from either the forward or reverse strand of the original dsDNA fragment.
From there it’s obvious that you can use this information to filter out errors that occurred during amplification (including bridge amplification):
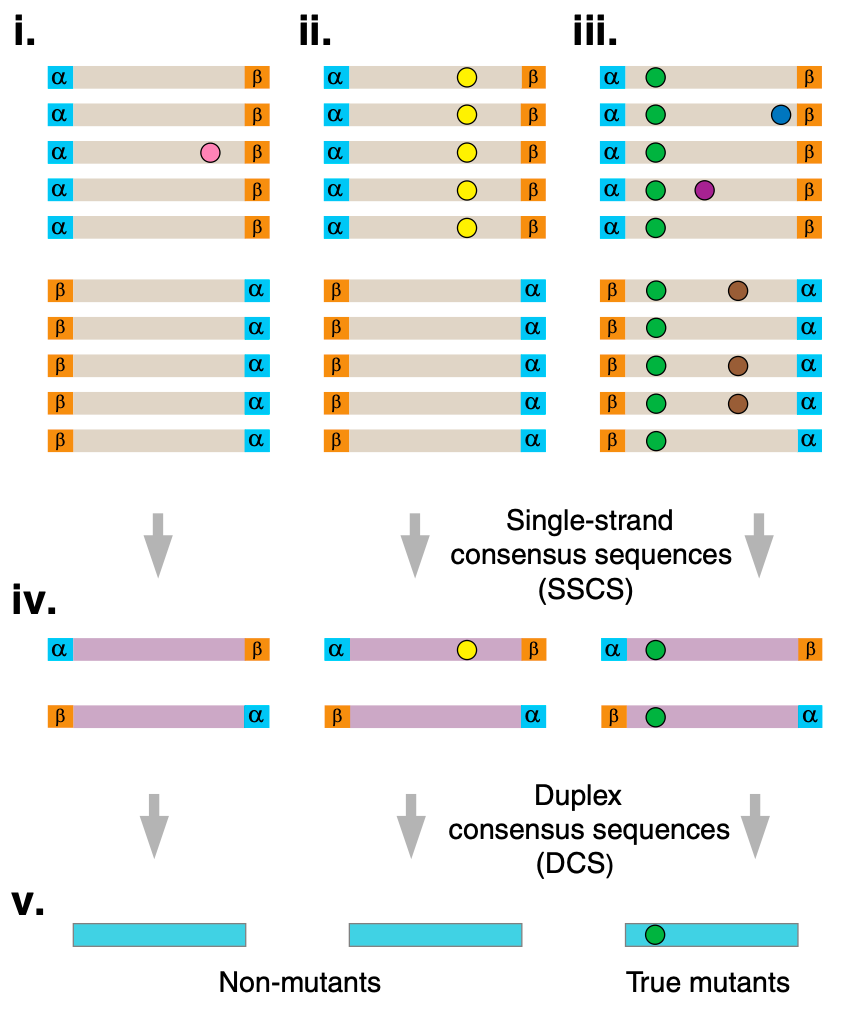
For amplification errors to propagate they’d need to occur at the same position, and of the same base. So, I’d assume a ballpark estimate is somewhere around Q60… and their reports include identifying mutation frequency down to a rate of 10^-5.
Problems
Wow, great! Q60 reads, who wouldn’t want that!
Well the major problem is that you’re going to throw away a lot of throughput. At best you will need to sequence each strand 2 to 4 times. This might be fine if you have an amplification step in your protocol anyway. Much like UMIs the Twinstrand process will just provide additional information removing error and bias.
But unlike UMIs you want to optimize for duplicates. And not just duplicates but duplicating starting material a fixed number of times. I.e. the ideal is probably to see ~4 different sequences for every original fragment of dsDNA (one of each type).
In practice, this is problematic, in their patent they state “3.1% of the tags had a matching partner present in the library, resulting in 2.9 million nucleotides of sequence data”. As far as I can tell the input datasets was 390Mb of sequence data. Processed, corrected reads therefore represent about 0.75% of the input dataset. This is a huge hit of your throughput.
The above describes the original IP, from ~2012. Most of their patents appear to be based around this basic process. However a patent from 2018 looks like it might be worth digging into in more detail. In this patent it looks like they try to more closely model errors that occur during the sequencing process (incorporating fluorescence intensity information into a two pass basecalling process).