


A GRAIL cfDNA Paper
I’ve been looking through some of GRAILs early stage cancer detection papers (I do some work in cancer diagnostics). This paper in particular from the end of last year helped round out the GRAIL story for me.
Previously my assumption on GRAILs development went something like this: GRAIL initially tried super high depth cfDNA sequencing. This kind of worked, but not very well. Then they added methylation into the mix and it started working much better. Along the way they tried out some other stuff (like cfRNA which I previously wrote about).
This paper was interesting to me because it expands somewhat on this story, and corrects a few misconceptions on my part.
The Datasets
Here they look at classification primarily based on 3 different sample types1:
WGBS: Whole genome sequencing of cfDNA methylation, 30x
SNV: Target sequencing of 507 Genes (3000x unique coverage)
WBC: Same as SNV, but for white blood cells.
Demographically the population seems well balanced, samples randomized to avoid batch effects etc. There were a total of 2800 participants (2261 analyzed, 1414 training, 847 validation), which seems like a pretty solid study size to me.
These primary datasets are further processed for use in training their classifiers:
SNV and SNV-WBC
Regular single nucleotide variants (SNVs) were called from the targeted sequencing dataset. But in addition to this they also filtered cfDNA SNVs using the white blood cell targeted sequencing dataset (SNV-WBC).
This, as the paper says “This compensated for normal within-person accumulation of somatic variants in the blood”.
WG Methylation
For the methylation work they do any targeting. So this is just 30x coverage across the entire genome using bisulfite sequencing.
But the methylation dataset is further filtered prior to classification. First they remove fragments that:
Occur at high frequency in non-cancer individuals2.
Have unstable methylation in non-cancer individuals.
Then from this dataset they retain only those fragments that:
Have at least 5 CpGs covered.
Average methylation ≥0.9 (hyper-methylated) or
Average methylation ≤0.1 (hypo-methylated)
They call this filtered set of reads UFXMs (Unusual Fragments of eXtreme Methylation). This results in only ~3000 fragments.
But that’s not all! They then look at where UFXMs lie on the genome, across cancer and non-cancer samples. This is then used to rank UFXM fragments from individuals.
The 7 UFXMs with the highest scores (for hyper and hypo-methylated fragments) are then selected. These 14 fragments are used to create features for use in the classifier!
If I’m reading this right, that’s something like 600 million filtered down to 14 reads?!
Classification
I’m not actually super-interested in evaluating classification performance from this paper. While this is part of the evolution of the Galleri test, the results here probably don’t reflect the classification performance of Galleri (more on that later).
Nor am I really interested in the details of the training and classification process beyond what datasets were used. I’m more interested in the general trend we see in classification performance, and how this has influenced further work.
This (adapted) figure sums it up for me, and was rather surprising:
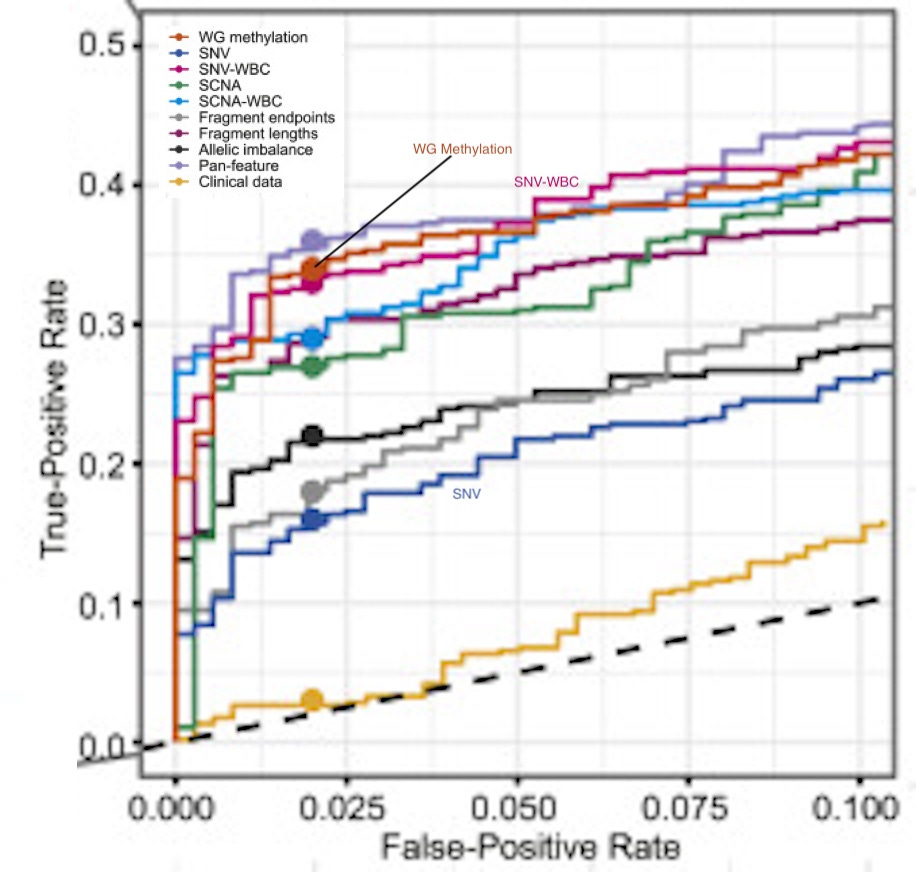
The figure includes a bunch of other approaches they tried (CNVs etc.). But I’m only interested SNV, SNV-WBC and WG methylation.
Using single nucleotide variants (SNVs) alone sucked! The classification performance is the worst of all the methods tried. But suddenly when you remove the white blood cell signal (SNV-WBC) it’s one of the best performing classifiers!
Removing background noise is really really important!
Whole genome methylation performs a little better than SNV-WBC. But perhaps more importantly, WG methylation was also significantly better at determining cancer signal origin:
“Among the three CSO classifiers, the WG methylation CSO classifier accurately predicted CSO for 75% (95/127) of jointly detected cancer samples, whereas SCNA and SNV-WBC accurately predicted CSO for 41% (52/127) and 35% (44/127) of cancer samples”

This is all the more impressive when you consider that the WG methylation classifier used an untargeted dataset. Of course the natural progression of this work is therefore to build a targeted methylation based test… which is exactly what they did to develop Galleri:
“These results informed the design and performance of the recently reported targeted-methylation-based cfDNA MCED test, which showed substantial improvements relative to the top-performing prototype tests evaluated here and formed the basis for the Galleri® MCED test.”
Using targeting they can get much higher coverage on the methylation regions of interest. This likely improves performance significantly, and if I have a chance I’ll review one of the targeted methylation papers in the future.
But for now, you can find a few more brief thoughts below!