
Are long reads useful for infectious disease testing?
I’ve been trying to answer the following question for a few months now:
In a SARS-CoV-2 sample, what is the SARS-CoV-2 fragment length distribution?
Finding a solid, or even approximate, answer to this question seems surprisingly hard.
But in this post, I’m going to try and puzzle this out a bit. The dataset I’d ideally like to see is really an SARS-CoV-2 anterior nasal or nasopharyngeal meta-genomic sequencing dataset using a long read platform.
But either these datasets don’t exist, the data is not available, or I’ve simply not been unable to find them.
The majority of SARS-CoV-2 in a sample is not viable
It’s been suggested that SARS-CoV-2 in saliva has a half-life as low as 30 minutes. This means that viral envelope degrades pretty quickly and it seems reasonable to expect that SARS-CoV-2 genomes might be heavily fragmented.
There are studies that have used approaches to inhibit amplification of virus’ which lack interacted membranes. One such approach is the use of PtCl4. This binds to nucleic acids and apparently inhibits amplification:

This paper used the approach to measure only the viable SARS-CoV-2 virons in a sample. They found that when comparing regular Ct values to “active virus only” Cts values dropped approximately 5 points:
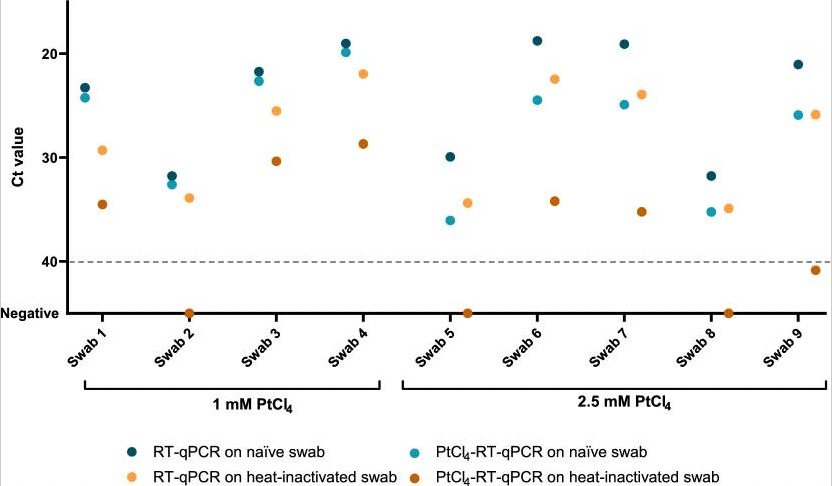
A decrease of 5 Ct points seems to roughly correlate to a 10 to 100 fold decrease in viral load based on other reports. This isn’t a particularly quantitive result. But it suggests to me that roughly an order of magnitude more inactive (and likely fragmented) SARS-CoV-2 is present in a sample than complete intact viruses.
A sequencing based study I found also suggested that SARS-CoV-2 is heavily fragmented in most cases. This was an amplicon based study, but one where’d they’d made an effort to calculate a “viral fragmentation score”. They also found that SARS-CoV-2 was heavily fragmented, and that fragment size was related to disease progress:

This isn’t a bad thing…
While the above reports have their limitations, they suggest that SARS-CoV-2 is heavily fragmented in most samples. This would mean much of the signal in a qPCR test is coming for non-infectious material.
This isn’t a bad thing in my view. The presence of not yet completely degraded SARS-CoV-2 RNA (or sgRNA) is being used as a proxy for the presence of active viral material. Given how quickly the material degrades seems perfectly reasonable.
…but their are implications for sequencing
This has a few implications for sequencing. Firstly, it suggests that if you are performing long read meta-genomic sequencing of infectious diseases and expecting to see lots of complete genomes you may be disappointed. No matter how much you optimize the sample prep, the input material is likely just heavily degraded.
In addition to this, your options in terms of enriching viral material solely based on fragment length may be limited. Human derived material in many samples seems to be of a similar fragment size (and cell-free).
You might just want to discard all non-viable SAR-CoV-2 virons. This might well work, but in diagnostic content (which is one of the things I’m interested in) may well result in a lower detection limit…
On a positive note, if qPCR is often detecting a virus based on degraded material, NGS may have a sensitivity edge over qPCR. When a few viral fragments are present, and they do not contain the qPCR target they will be missed in a qPCR assay. However NGS will be able to identify the source of any viral fragment sequenced.
What do you think?
I’ve been surprised that I’ve not been able to find a clear and concise answer to this question, and it’s entirely possible I’ve just been looking in all the wrong places. So if you think you might have the answer to this question, or know where it might be found, please get in touch (new@sgenomics.org, in the comments, or on twitter)!