
Modeling Sequencing Sensitivity
In a previous post I discussed the sensitivity of sequencing and how, in the limit, sequencing is a more sensitive technique than qPCR. In this post I’m going to discuss a couple of models I put together to try and explore this further.
First, let’s briefly review the model from the previous post. In this model we assume there is a single viral/target genome fragment (SARS-CoV-2: 30Kb) in our sample, of a given fragment size. The qPCR column shows the probably that this single fragment will cover the amplification region.
The sequencing columns show the number of fragments you would expect to see in a given set of reads.

This hopefully expresses why sequencing is potentially more sensitive than qPCR. But I decided it would be interesting to look at the problem from the opposite direction.
So, I put together a second model. In this model I’m looking at varying target fraction, modeling sequencing and qPCR output using a number of input parameters.
Specifically here our parameters are:
Total RNA: 10ng
Genome Size: 30Kb
rRNA fraction: 60%
qPCR Target Region: 90nt
Fragment Size: 1000nt
Read Count: 40M
I’ve struggled to find good numbers of the total RNA yield from an NP swab, and may revisit this when I’ve finished reviewing Metagenomics sequencing papers. The 1ng used in the previous model could be an under-estimate and might bias the model toward sequencing, which is why I’ve used 10ng here.
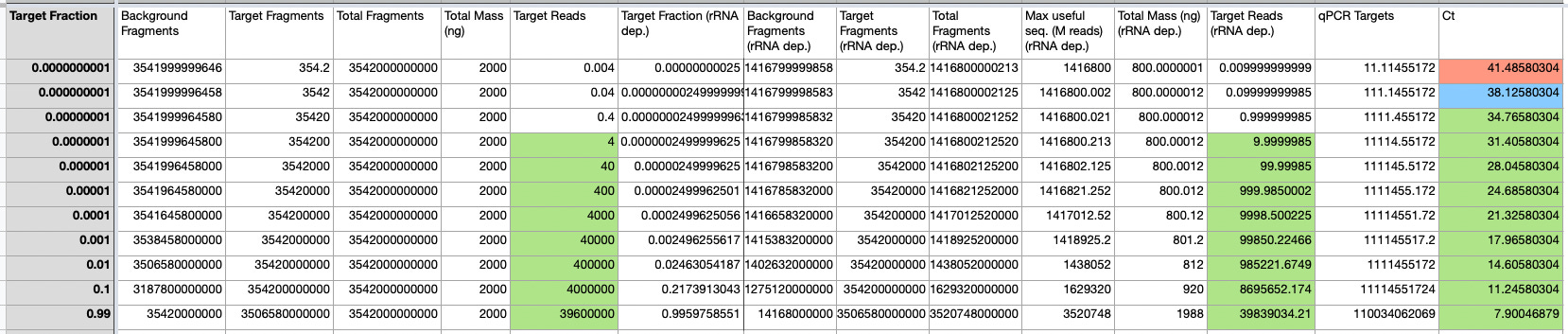
The parameters above roughly match the JumpCode paper I previously discussed. Throwing all these numbers into the modeling sheet gives us estimated read counts and Ct values for qPCR. Ct’s are estimated from standard curves and the number of fragments we would expect to be “on target”:

The model suggests that using the inputs above we expect to hit a sensitivity of ~Ct 35 with sequencing, which is also roughly the conclusion the JumpCode paper comes to.
With these parameters the model output is in reasonable agreement with the JumpCode dataset:
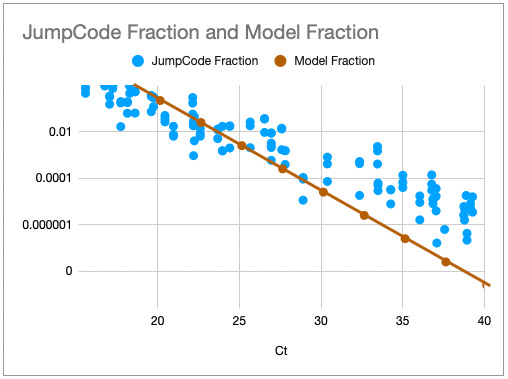
We can push the model is various directions, for example if we have 2000ng of input material, qPCR starts to out-perform sequencing:
But these results then no longer match the JumpCode dataset as well:

Results are similarly poor if we increase the fragment size to nearly cover the whole genome. You should be able to take a copy of the spreadsheet and play with it, pushing the models in various directions. Comments and suggestions are most welcome!
I’ll be revisiting this looking at datasets from other papers, so you can expect a follow up to this at some point in the future.
Thank you for the article. Just wanted to comment quickly to help your analysis. If you are on the hunt for sensitivity, you would do ddPCR or smFISH, not qPCR. Both method have single molecule sensitivity given you are able to extract a single molecule of viral nucleic acid from a nasal swab. The efficiency of the extraction itself is highly variable depending on how degraded it is, time since isolation, storage buffer, etc but is independent of the detection method.
qPCR is just quick and easy, that's why it's popular. No need to mess with a keyboard. Sensitivity is not really the point there, usually.