
Sequencing: More sensitive than qPCR
qPCR is widely believed to be the “gold standard” in molecular diagnostic testing. In this article I’m going to make the argument that not only does meta-genomic sequencing have broader utility than qPCR allowing the detection of known and unknown viruses, but that it’s also more sensitive.
qPCR has a very low LoD (limit of detection), in some cases a handful of copies can be detected in a sample. This is generally assess using synthetic material, spiked into a sample to create standard curves like the following:

Because qPCR targets a particular region, it’s relatively insensitive to background material. That is, you can have human material (mostly rRNA in the case of RT-qPCR), bacterial material or whatever else floating around, and it’s not going to cause much of an issue.
Meta-genomic sequencing however, is sensitive to background material. To ensure that your target(s) of interest are captured, you also need to sequencing though all the background fragments.
At first glance, it seems like forgone conclusion. qPCR will alway be more sensitive than sequencing. In practice however it turns out that this may not to be the case…
In Practice Sequencing Performs Better
While qPCR should be more sensitive in theory, a number of publications have shown that meta-genomic sequencing is more sensitive than PCR. In many cases, sequencing was able to detect low quantities of SARS-CoV-2 material in patients that had tested negative using qPCR.
In one study sequencing data suggested that “13% of PCR-negative samples were false negatives”. Another found that sequencing (46.7M reads/sample) was more sensitive than a PCR Panel, and in one case recovered a complete genome with 233x coverage which was not detected by the panel.
Yet another study showed (15M reads/sample) that sequencing out-performed a PCR panel:
“Untargeted meta-genomics detected 12 additional respiratory viruses, 3 of which were targeted by the RVP. Seven of the 12 additional viruses (58.3%) were confirmed by qPCR, 3 were qPCR negative, and 2 could not be tested due to due to limited sample volumes. The 3 viruses detected by untargeted metagenomics but not qPCR had low viral read counts (4 to 31 reads). However, these reads were located across unique regions of viral genomes and sequences differed by several nucleotides from those of other viral strains detected at higher read counts within the batch, suggesting that they were not misclassifications and unlikely to be contaminants.”
In Theory Sequencing Performs Better Too!
Given that the LoD for qPCR should be very low, why do we find that the performance is worse than sequencing in the above studies?
There are several likely reasons, and I’ll address them one at a time below, but at a high level qPCR needs a small number of target regions to be fully intact to enable detection. Sequencing by its nature, addresses the entire genome any short (20bp+) read will uniquely identify a viral genome.
Mutations
Single mutations in primer sites can result in PCR failure (no amplification). Such mutations would have no effect on sequencing, where reads are in general sufficiently long to align with multiple mutations.
I suspect mutations are not the most significant issue in the majority of cases, but it is an issue worth keeping in mind, as viral genomes evolve and potentially evade detection.
Fragmentation - Handling
The CDC SARS-CoV-2 Multiplex assay primers are about 90nt apart. qPCR is therefore able to detect SARS-CoV-2 if this 90nt region is present in the sample. SARS-CoV-2 is a 30Kb genome. And as we’ve seen with long read sequencing platforms, keeping 30Kb+ fragments fully intact often requires special handling. If during sample prep the SARS-CoV-2 genome is fragmented anywhere in that 90nt region, amplification will fail. This is unlikely to be significant at high viral load, but when single digit copies are present in a sample, it could cause issues.
Fragmentation - At Collection
Beyond this, I suspect SARS-CoV-2 is fragmented in the sample as collected. I’ve previously looked at how quickly live/viable SARS-CoV-2 degrades in a sample, these results suggest that SARS-CoV-2 degrades relatively quickly, and samples may therefore contain significant quantities of fragmented cell-free SARS-CoV-2 RNA.
Other reports suggest that CT values of SARS-CoV-2 in viral transport medium, increase by 3 points over a 14 day period, an approximately 10-fold reduction. This also suggests to me that it’s likely live virus is constantly degrading in the body and after collection. A significant fraction of the nucleic acid material used by qPCR and sequencing may therefore be fragmented.
Yet other reports suggest that samples from high CT “disease resolved” patients are likely “residual noninfectious fragmented viral RNA being released as a result of immune clearance”. Samples taken 8 days after symptoms onset often fail to culture, indicating live virus is not present. Again, this all suggests to me that fragmented viral RNA is generated during the course of an infection, and remains present for sometime.
Elsewhere, sequencing was used to confirm this and suggested that in high CT patients viral RNA was fragmented and non-viable:
“Bioinformatic reconstruction of sequenced RNA showed how samples did not have the whole viral genome, but only few and very short gene fragments (<600 nucleotides). This finding indicates that the residual RNA detected by molecular testing was not functional and represented only pieces of the degraded viral genome.”
Fragmentation - sgRNA
During the SARS-CoV-2 replication cycle, sgRNA is created. This sgRNA is also available to a meta-genomic sequencing based diagnostics, giving us an edge over qPCR. The presence of sgRNA (which is cleared relatively quickly) also indicates that replication is actively taking place.
One study looked at 2 sgRNA regions, and found that they represented about 20% of the ORF1ab counts. There are 10 canonical RNAs in SARS-CoV-2. It therefore seems possible that the total quantity of sgRNA could be significant in infectious individuals.
Effect of fragmentation
If viral genomes are fragmented it puts qPCR at a significant disadvantage. While sequencing will be able to read any fragment, align it to a reference and detect the presence of a virus, qPCR will only be able to “use” those fragments spanning the amplification region.
To put some numbers on this, let’s assume we have roughly 1 nanogram of RNA1. Running the numbers gives us some sense of where and why sequencing out-performs qPCR. In the extreme, if we assume a single fragment of SARS-CoV-2 RNA is present in our sample with an average fragment length of 200bp there's a probability of 0.0037 (~0.04%) that any given fragment contains the amplification region. If qPCR and a LoD allowing it to detect a single fragment, it would outperform untargetted meta-genomic sequencing approaches using <10 million reads. With depletion of human material (assuming human/rRNA is ~50% of material) we can approach qPCR sensitivity using as few as 20 million reads.
More realistically the qPCR LoD is at least 5 copies. In this scenario, we need 5 fragments which cover the amplification region. Here, sequencing will out-perform qPCR in all cases.
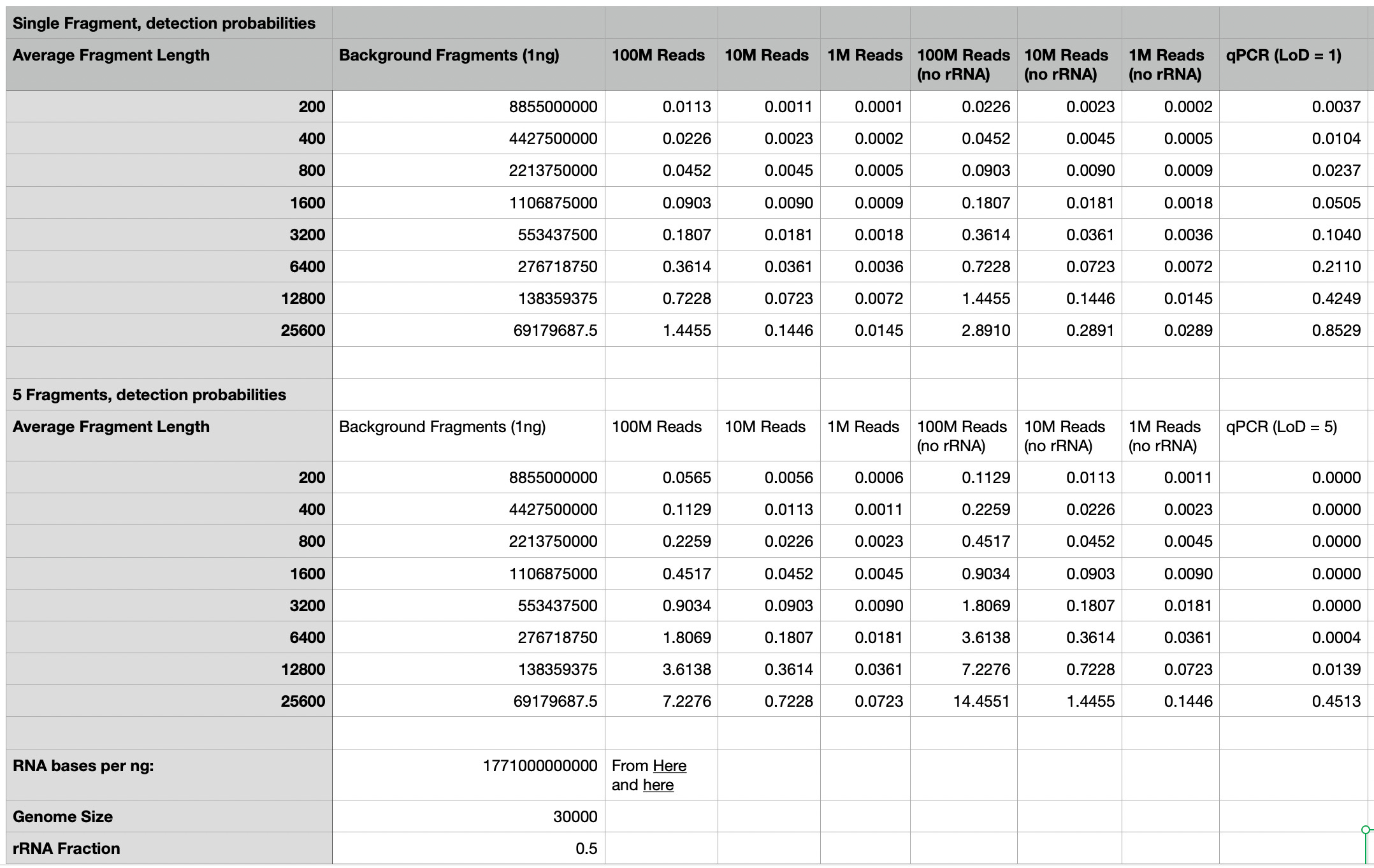
This is an obviously incomplete model (and assumes that both the target genome, and background material have the same fragment size), but what I’m trying to convey is that the sensitivity of qPCR, as compared to sequencing is likely strongly related to the fragmentation level of both the target and the background material.
This may go someway to explaining why practical implementations of meta-genomic sequencing have exhibited higher sensitivity than qPCR. It also gives us hope that a meta-genomic sequencing based diagnostic platform, might not just out-perform qPCR in terms of providing broader detection, but also in terms of sensitivity using relatively modest read counts.
typically yields seem be between 1 and 10 nanograms. Exact values are hard to find. From what I can tell many authors consider the Qubit to be a magical device used to bless samples the primary function of which is to allow you to state that “samples were quantified using a Qubit Fluorometer” in your publications.