


Oxford Nanopore Error Rate Is >5%?
UPDATE: You should probably checkout the followup post on this analysis here.
A friend of mine is building a genomics analysis platform called GenomeMiner.ai. Eli and co. have been building this platform to support their own internal microbe development pipeline and are expanding this to support external users. If you’re looking for user friendly cloud computing interface to private and public Bioinformatics tool, you should check it out1.
Anyway! I’ve been using it to align and play with some ONT data to help debug the platform… I also would kind of like to know what the overall error rate of Oxford Nanopore data is…
ONT will provide all kinds of weird error metrics2… anything other than the overall per-base accuracy. To find this, you have to align the data yourself.
So we went and grabbed some public ONT data3 , the best studied genome at present seems to be HG002. So we should be able to expand this analysis to multiple ONT flowcell revisions as well as PacBio and Illumina data.
So I grabbed what is hopefully a reasonable reference for HG002. And we basecalled and aligned4 a subset of the reads against the maternal, paternal and a combined reference5 the empirical quality versus true quality as calculated using BEST6 looks like this:

Quality scores seem kind of somewhat reasonably well calibrated up to about Q20. Beyond this empirical and fastq quality diverge significantly. Using the combined reference helps here.
However, the actual population of bases above empirical Q15 is pretty much zero:

Which is reflected in the overall identity statistics:
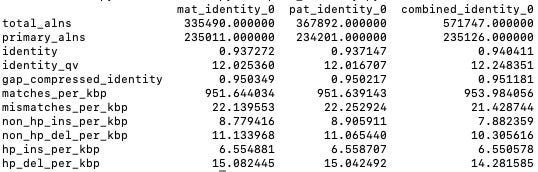
My reading puts the error rate somewhere north of 5% if we include mismatches and indels. Breaking this down further… if I’ve understood correctly
We have homopolymers (hp) indels totaling 20.83 per 1000. Non-homopolymer indels of 18.18 per 1000. Mismatches of 21.42 per 1000. Adding this all up (which probably isn’t quite the right thing to do) gives us a totally error rate of 6.04%7
So for this R10.4.1 Simplex dataset is representative, and there isn’t anything wrong with this quickly thrown together analysis I can’t quite see how these can be called Q20+ reads. Given that at >1% error rate, they would be significantly lower accuracy than the first generation Solexa sequencers.
That’s no problem! Nanopore sequencing has a number of advantages in other respects (longest available reads, direct RNA). But…
Calling them Q20 based on Oxford Nanopore often quoted per-read modal accuracy… that…. well that would be what we call in the business NumberWang.
I’d welcome comments and suggestions on what might be wrong with this analysis and improvements. I’ll likely try and grab a bunch of other datasets to expand the analysis and will incorporate any suggestions there. Feel free to ping me via email (new@whitefordresearch.com) or on the Discord.
And subscribe! I’m still bitter about Century of Bio!
I have no vested interested here, but Eli is a nice guy and GenomeMiner is neat. The ideal use case is probably an early stage startup that needs to migrate their tools away from an academic cluster and provide a user friendly interface to support researchers in an industrial context.
Mostly they just say things like “Q20” reads. But the fine print will state things like “98.3% modal (Q18, simplex)”. When you’re per-read accuracy distribution isn’t gaussian this isn’t easily comparable with a “Q20 average” on another platform (the original Genome Analyzer was ~Q20 average).
Reference here: https://labs.epi2me.io/giab-2023.05/
s3://ont-open-data/giab_2023.05/flowcells/hg002/20230429_1600_2E_PAO83395_124388f5
From memory we re-basecalled this using the dna_r10.4.1_e8.2_400bps_fast@v4.2.0 model. The subset we took resulted in a ~5Gb BAM.
Using Dorado Align
I just concatenated the maternal and paternal references…
It’s a great tool, but the name makes it almost un-googlable.
Looking at it in the other direction the numbers seem to add up. If we take the matches (932.98 per 1000) we get an error rate of 4.6%. I assume this doesn’t take into account deletions which come to 2.4% giving 7%.
Interesting Nava, thanks a lot for this. I've always been puzzled why ONT isn't wiping out competitors if their accuracy is as good as the impression conveyed, in addition to their low capex, portability etc; I guess your analysis explains it.
How much improvement would ONT's duplex reads make here? IIRC they claim it brings them to Q29 modal.
Way easier said than done, but it'd be great to have SBS reads aligned to the same reference for comparison...