
A Look At Some PacBio Subreads
To review: PacBio uses a process called CCS (cyclic consensus sequencing). Each DNA fragment is read multiple times. This resolves a fundamental issue with many single molecule sequencing approaches: high error rates. By observing the same molecule multiple times, you can correct errors and create a highly accurate “consensus” read.
Over on the Discord someone pointed me toward a raw subread dataset. From the metadata this dataset appears to come from a Sequel II (or IIe?) and HG002. Looking at the headers also confirmed that the data appears to be sampled at 100Hz (confirming another suspicion from the previous post).
I decided to have a quick poke around…
First I used the PacBio tools to generate CCS reads… the report generated here was interesting in itself. Only 36% of the 10000 read sets passed filters, the majority failed due to “Lacking full passes”, documentation defines this as “There were not enough subreads that had an adapter at the start and end of the subread”.
This was surprising to me. On the Revio of 25M wells, ~6M produce HiFi reads: ~25%. This suggests that 30% of wells produce no/invalid data. The other 70% produce some kind of read. And ~35% of these produce HiFi reads:
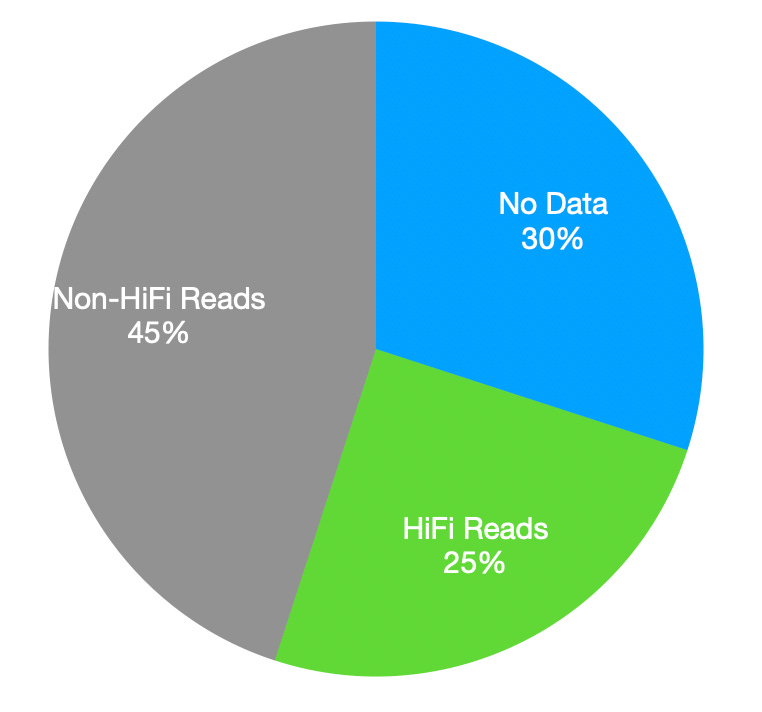
That’s a lot of data to throw out1 and a bit lower than my previous estimates. It also suggests a lot of room from improvement if you can improve the HiFi yield or underlying read accuracy!
I thought it might be interesting to look at the subread length distribution. So I plotted the subread length as a fraction of the final consensus read length. I expected the subreads to be shorter than the final generated read but this turns out not to be the case. Subreads are generally a little longer than the final consensus:
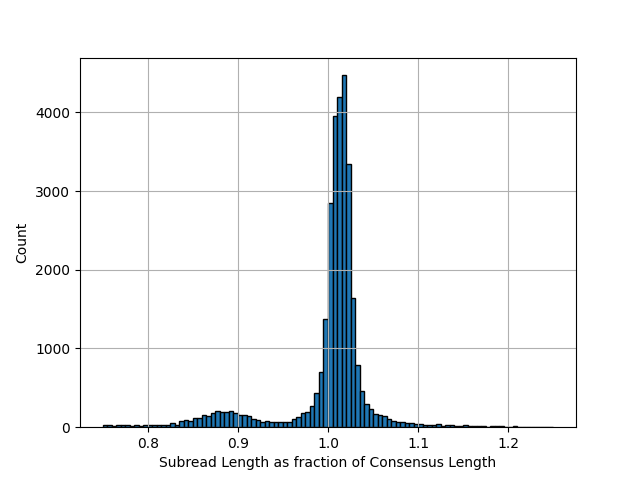
I assume this is due to trimming the ends of the reads to remove low coverage/quality bases. This is makes sense and is a relatively easy thing you can do to improve accuracy.
The CCS process took ~3mins on my very old 48 core Xeon(R) CPU E5-2697 2.7GHz. That suggests ~30h for a full Revio run. I’d expect a modern system to be at least twice as fast (15h). I’d also hope that you can run at least part of this process in parallel with sequencing…
What was the accuracy of the CCS calls? (without DeepConsensus)
Well… the alignment is currently running. So stay tuned for those results!
Scripts
Installed tools, grabbed data with:
conda install -c bioconda pbbam pbccs actc
gsutil cp gs://brain-genomics-public/research/deepconsensus/quickstart/v1.2/n10000.subreads.bam .Datasets
#/bin/bash
samtools view n10000.subreads.bam > n10000.subreads.sam
cat n10000.subreads.sam | awk '{print $1 " " length($10)}' > n10000.subreads.lens
cat n10000.subreads.lens | awk 'BEGIN{FS="/"}{print $1 "/" $2 " " $3}' | awk '{print $1 " " $3}' > n10000.subreads.lens.id
./doccs
cat 10000-of-00500.ccs.sam | awk '{print $1 " " length($10)}' > 10000-of-00500.ccs.sam.len
cat 10000-of-00500.ccs.sam.len | awk 'BEGIN{FS="/"}{print $1 "/" $2 " " $3}' | awk '{print $1 " " $3}' > 10000-of-00500.ccs.sam.len.id
sort 10000-of-00500.ccs.sam.len.id > 10000-of-00500.ccs.sam.len.id.sort
sort n10000.subreads.lens.id > n10000.subreads.lens.id.sort
join n10000.subreads.lens.id.sort 10000-of-00500.ccs.sam.len.id.sort > n10000.subreads.joineddoccs
#/bin/bash
n=10000 # Set this to the shard you would like to process.
n_total=500 # For a full dataset, set to a larger number such as 500.
function to_shard_id {
# ${1}: n=1-based counter
# ${2}: n_total=1-based count
echo "$( printf %05g "${1}")-of-$(printf "%05g" "${2}")"
}
shard_id="$(to_shard_id "${n}" "${n_total}")"
echo "$(nproc)"
# --chunk="${n}"/"${n_total}" \
ccs --min-rq=0.88 \
-j "$(nproc)" \
n10000.subreads.bam \
"${shard_id}.ccs.bam"Generate Histogram:
import matplotlib.pyplot as plt
# Read the data from the text file
with open('n10000.subreads.joined.frac', 'r') as file:
lines = file.readlines()
# Extract the second values and convert them to floats
values = [float(line.split()[1]) for line in lines]
# Plot the histogram
plt.hist(values, bins=100, range=(0.75, 1.25), edgecolor='black')
plt.xlabel('Subread Length as fraction of Consensus Length')
plt.ylabel('Count')
plt.grid(True)
# Save the plot as a PNG file
plt.savefig('histogram.png')Alignment:
minimap2 -t 10 -x map-pb /mnt/sdb1/HGP002/HG002.combined.fa.gz ./10000-of-00500.ccs.bam -o 10000-of-00500.ccs.bam.bamBut a reasonable business decision that was also made on the original Illumina instruments which threw out a large fraction of likely highly errored reads.