


Illumina Complete Long reads - synthetic not synthetic long read technology?
So at AGBT more was disclosed about Illumina’s long read approach, which they now call “Illumina Complete Long Reads” and originally called “Infinity”.
I wrote about Infinity when it was first announced, and I think most of what I said still holds true (I just briefly looked over the AGBT slides posted on twitter, which you can find in the Discord, nearly 200 people there now!).
In September, DeSouza then made the following quite confusing statement about Infinity:
“Illumina Complete Long Reads you know that as Infinity meets this need. This technology gives you long read capabilities without compromise. And without needing a new platform. Illumina Complete Long reads is not synthetic long read technology. With Strobe reads and difficult work flows and complex informatics. It is a complete and accurate representation of the genome at the single molecule level this means you can get contiguous long reads to close key gaps. That’s a game changer.” - Illumina’s Innovation Roadmap September 28th 2022
Based on what we now know this statement seems at best very very misleading. I have a hard time figuring out how to characterize the Illumina approach as anything but synthetic long reads.
Very disappointing. And as other recent announcements. Boring.
Back in March of last year I posted an approach that I thought was… at least more entertaining. But it was behind a paywall. Here it is now for you all.
I’ve just for fun filed a patent on the approach under Reticula. And it could integrate pretty well into that platform.
Background - Clusters and Phasing
The cluster based bridge amplification approach was originally proposed and used to provide more emitted signal than available with single molecule approaches. This has had many advantages in terms of SNR and allowed cheaper cameras/optics to be used.
The approach also meant that a certain amount of photo-damage could be tolerated. If a fraction of molecules (strands) within a cluster are photodamaged, the remaining molecules will still provide sufficient signal to allow sequencing to continue and the sequence to be determined.
But as we showed previously, the approach introduces phasing artifacts, and ultimately these limit read length. This isn’t the case with many non-amplified single molecule techniques, where we can often reach read lengths of 10Kb+.
The Approach
Can we somehow retain the advantages of clusters, but enable long reads?
One method would be to perform single molecule sequencing on the individual molecules in a cluster. In a sense, this is similar to PacBio’s “HiFi CCS” reads. We read each strand independently, and then combine the information from multiple strands to reconstruct the original template.
This wont work well on standard clusters, with diffraction limiting imaging resolution however. Clusters were originally ~1 micron, containing ~1000 clonal copies, a spacing of <30nm between individual molecules. Clusters on the Novaseq are even smaller (~200nm).
It might be possible to use a super-resolution technique to resolve individual molecules. But I suspect another approach might work better.

That is to say, we could “decimate” our clusters. That is stochastically deactivate strands in the cluster. We can do this by flowing in a mixture of natural and terminated (non-reversibly terminated) nucleotides. This gives us a larger distance between active strands in a cluster, such that we can resolve and sequence the individual strands.
Single molecule sequencing on each of the strands in a cluster can then proceed as normal using reversible terminators (or other methods). Once sequencing is complete a multiple alignment (similar to PacBio’s CCS) will generate a corrected consensus read for this cluster.
The process above decouples the amplification process from phasing issues. Phasing errors in one strand don’t effect the readout of an adjacent molecule. It’s a bit like being able to do PacBio CCS, but much faster, as you read all the strands in parallel.
The approach also gives us a couple of other potential advantages. Firstly, it could actually be beneficial to push strands further out of sync with each other. Let’s say you want to generate a 10Kb read, but you only want to run a 1000 cycle run (to keep the total run time short). You can obtain information different positions on each strand by pushing them out of sync. For example, by flowing in a mixture of reversibly terminated and unterminated nucleotides, to randomly stop extension at different points on the strand.
Beyond this, we may also be able to incorporate a “Twinstrand-like” error correction approaches. Our “single-molecule readout clusters” give us spacial information about polymerase errors, as they propagate from original template to other molecules in the cluster:
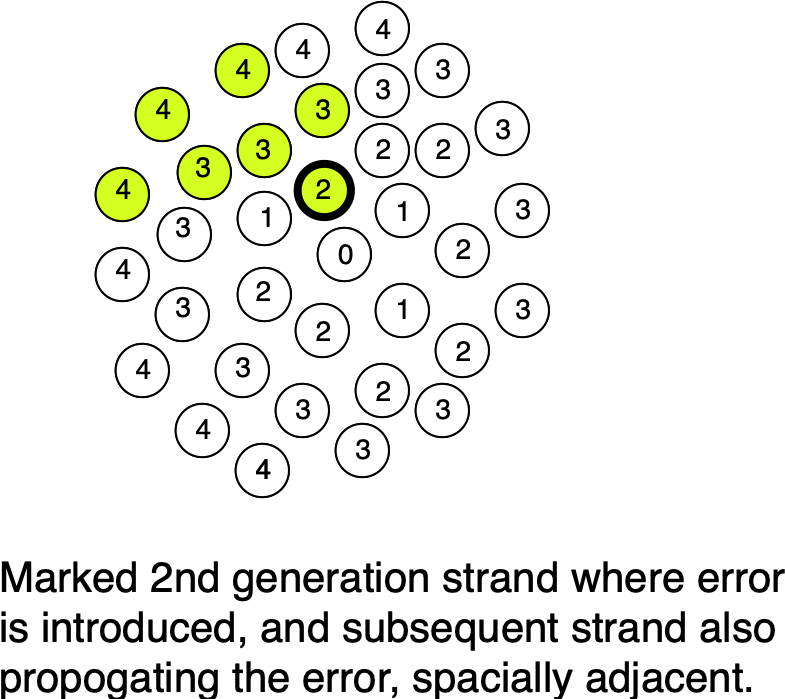
At worst we can easily identify those clusters where the consensus shows ambiguity and filter out these errors. In the best case scenario, we can actually model the likely “evolution” of these error’d strands in the cluster and correct the consensus.
An objection to this approach would be that you need a camera/optical system capable of single molecule imaging. The reality is that these have come down in cost significantly in recent years, a ~$10000 sCMOS camera will certainly work. But even low light consumer grade CMOS cameras should do the trick.
For the optical approach, you will likely need to use TIRF, this can get expensive with TIRF objectives costing >$5000. But prism, or other TIRF approaches would be cost effective.
I think this approach would be fun to try and it could potentially even integrate well in to some of Illumina’s current platforms.
If you’d like to chat about it send me an email (new@sgenomics.org) or ping me on Discord.