
Roche SBX (Axelios) Data Release
Roche have released data from Axelios! This takes the form of a bunch of BAM files. These are SBX duplex reads for GIAB samples. As such they should be comparable to widely available datasets from all major DNA sequencing platforms.
I decided to have a very quick, hacky, and possibly flawed poke around and grabbed the first part of the HG002 BAM1. Q scores are interesting, and appear to be distributed across 3 values. I believe the webinar may have more details here2. It would make sense if these Q scores represent the 3 classes of bases in this dataset:
Matching duplex bases (Q39)
Non-duplex bases (Q22)
Non-matching duplex bases (Q5)
The overall distribution of these Q scores also makes sense to me:
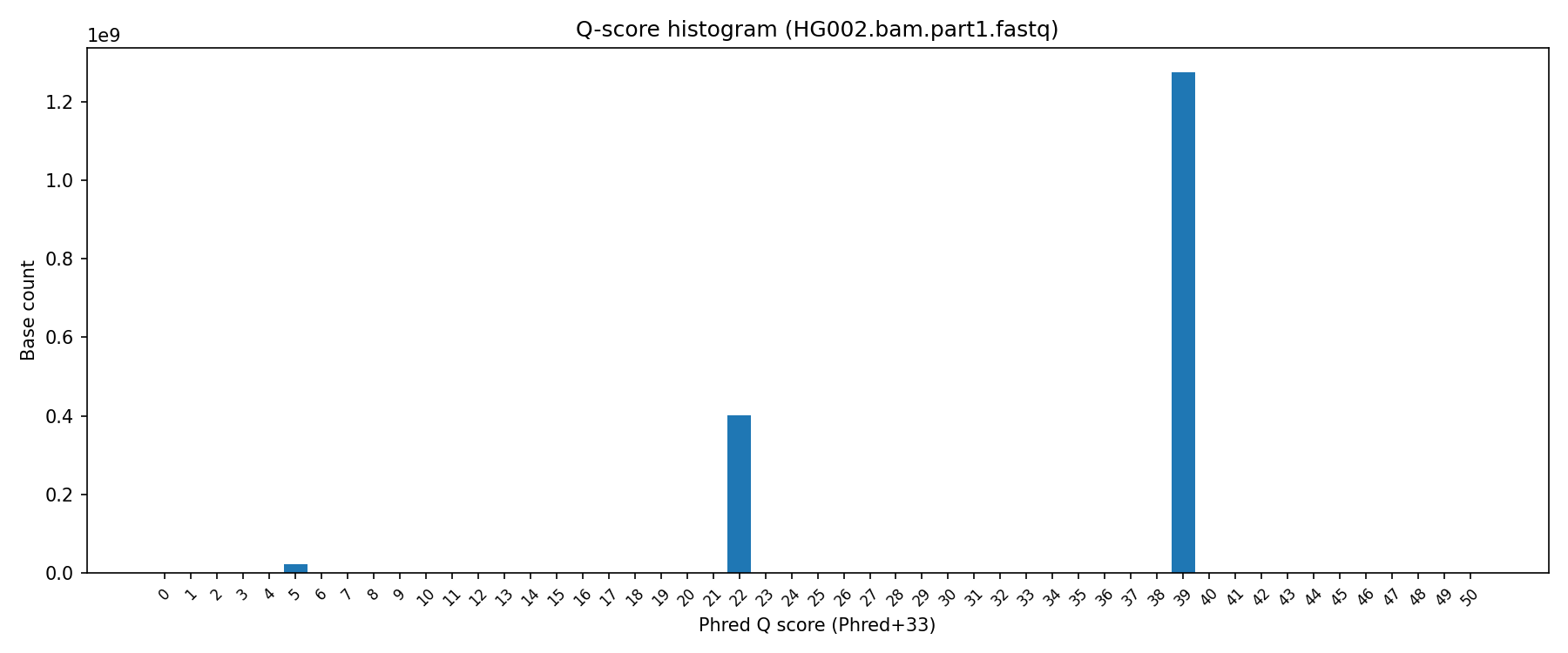
But the distribution based on read position… not so much:
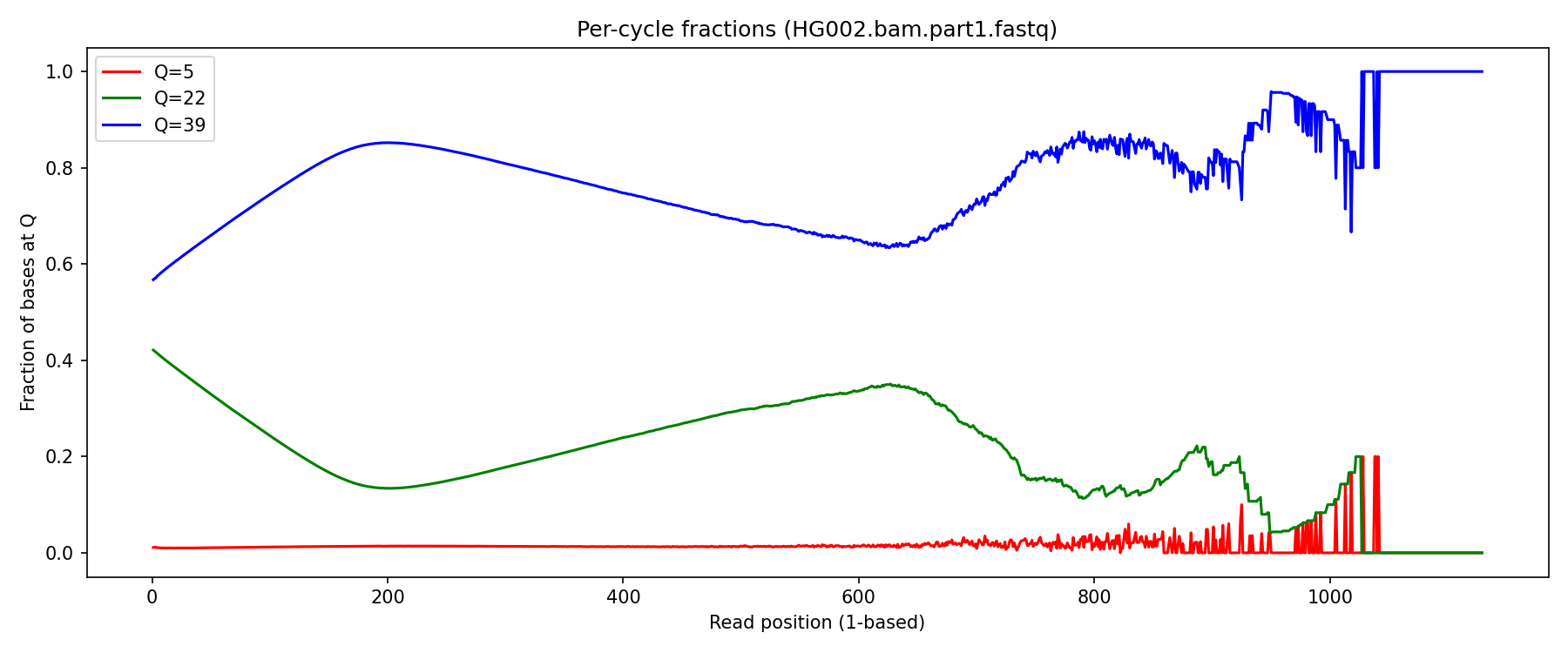
The Q5 distribution seems reasonable, it should be low and constant across the read. The noise toward the end of the read is likely in part due to only a small fraction of reads extending to this length.
However if you look at the absolute count of Q22 bases, they as expect sit at the beginning of the reads:
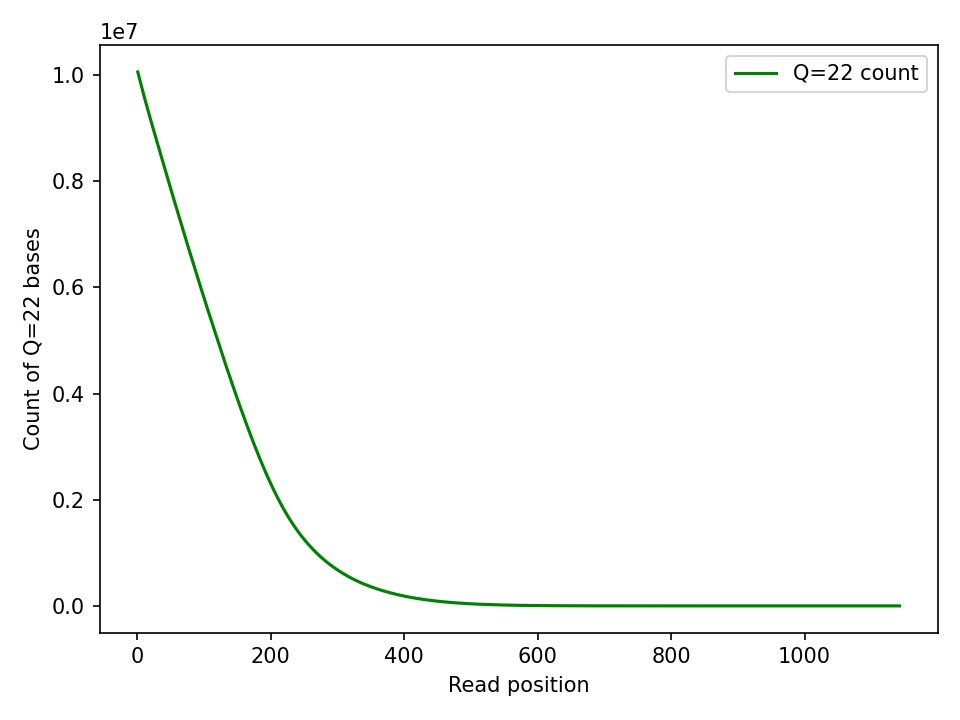
The absolute Q39 count peaks and then falls off. I assume this is a combination of partial read bases at the beginning of the read and read length falling off.
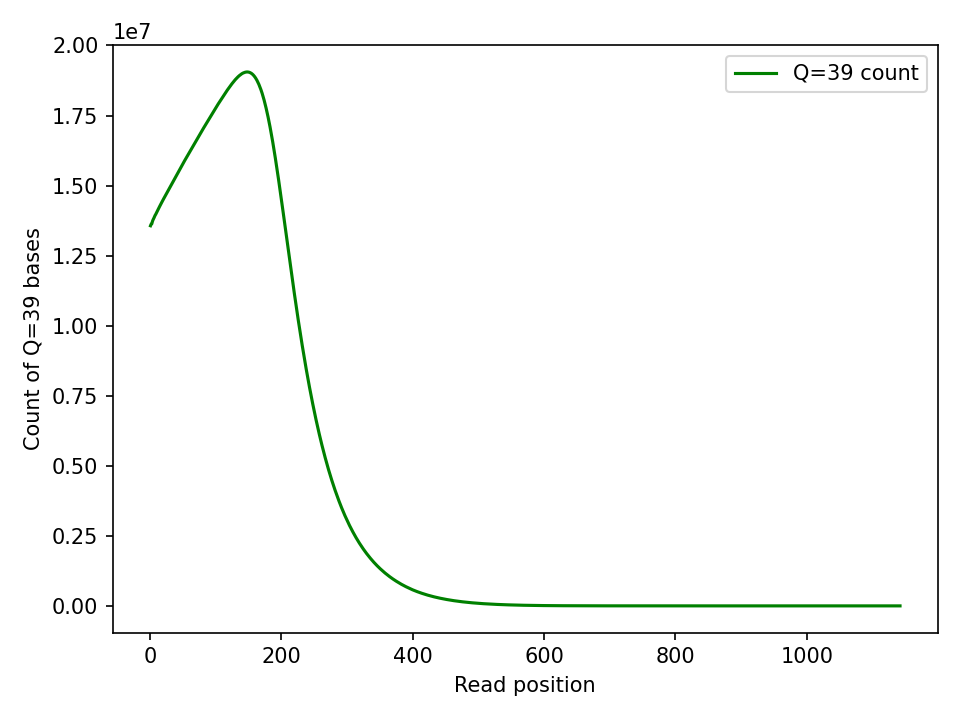
I think what’s happening here is that reads with partial bases are on average longer than those without. As read length extends the overall population of duplex versus partial falls off.
To verify this I calculated average read length for reads with and without partial basecalls very roughly. This gave me an average read length of 271.74bp for reads with leading non-duplex bases. And 219bp for reads that were full duplex3.
Perhaps worth a more systematic analysis at some point…
Summary
I’m just playing around at the moment. The dataset is duplex but contains simplex bases from partial reads, this should make it quite an interesting dataset to help us better understand the character of Axelios.
Adapting habits and methods to Axelios data is likely going to take more work than say transitioning from Illumina to MGI or Element.
But, it still seems hugely disruptive (pending pricing).
More in the SBX eBook!
There are many reasons why this might be a bad idea, including that these BAMs maybe sorted such that reads at the start of the file are not representative of the whole dataset.
I looked at the first 10Gb of fastq data generated from this file.
Some really rough and hacky scripts…
cat HG002.bam.part1.fastq| grep 777777 > 7s
cat HG002.bam.part1.fastq| grep HHHHHH | grep -v 777777 > N7s
new@MacBook-Air roche % python3 ./c.py 7s
File: 7s (Has 7s)
Total lines: 9810622
Average line length: 271.74
Lines < 400 chars: 9056661
Lines > 400 chars: 741965
new@MacBook-Air roche % python3 ./c.py N7s
File: N7s
Total lines: 14074151
Average line length: 219.05
Lines < 400 chars: 14059476
Lines > 400 chars: 13878