
The Utility of Long Reads...
Many years ago I wrote a paper called “An analysis of the feasibility of short read sequencing”. In this paper we looked at the percentage of substrings of each length that was unique with the human genome1. The idea was that if the majority of substrings of a given length were unique in the human genome it’s likely that reads of this length would be suitable for genome re-sequencing.
As it turned out, 25bp reads would allow you to address ~80% of the human genome with slowly diminishing returns after that:

From memory the Genome Analyzer launch spec was 25bp and I remember one advisor telling me that an investor2 had called him up wanted to know if our results supported the utility of short reads…
Now with the new telomere2telomere long read enabled references I figured it would be fun to try asking a similar question. What’s the utility of long reads?3 I.e. how much more of the genome to long reads let us address.
Back in the day running the above analysis was kind of a pain and required getting special access to a computer with a whopping 80Gb of RAM…4
I now have a server with 128Gb of RAM which is now essentially considered junk5. For various reasons the code now uses more RAM that it did back in 20056. As such I can only run the forward strand. But below is the uniqueness plot for T2T-CHM13v2.07. It should be noted that there seem to be a couple of issues8 with this code and as it’s the forward strand only the analysis is far from ideal… but might be instructive anyway:
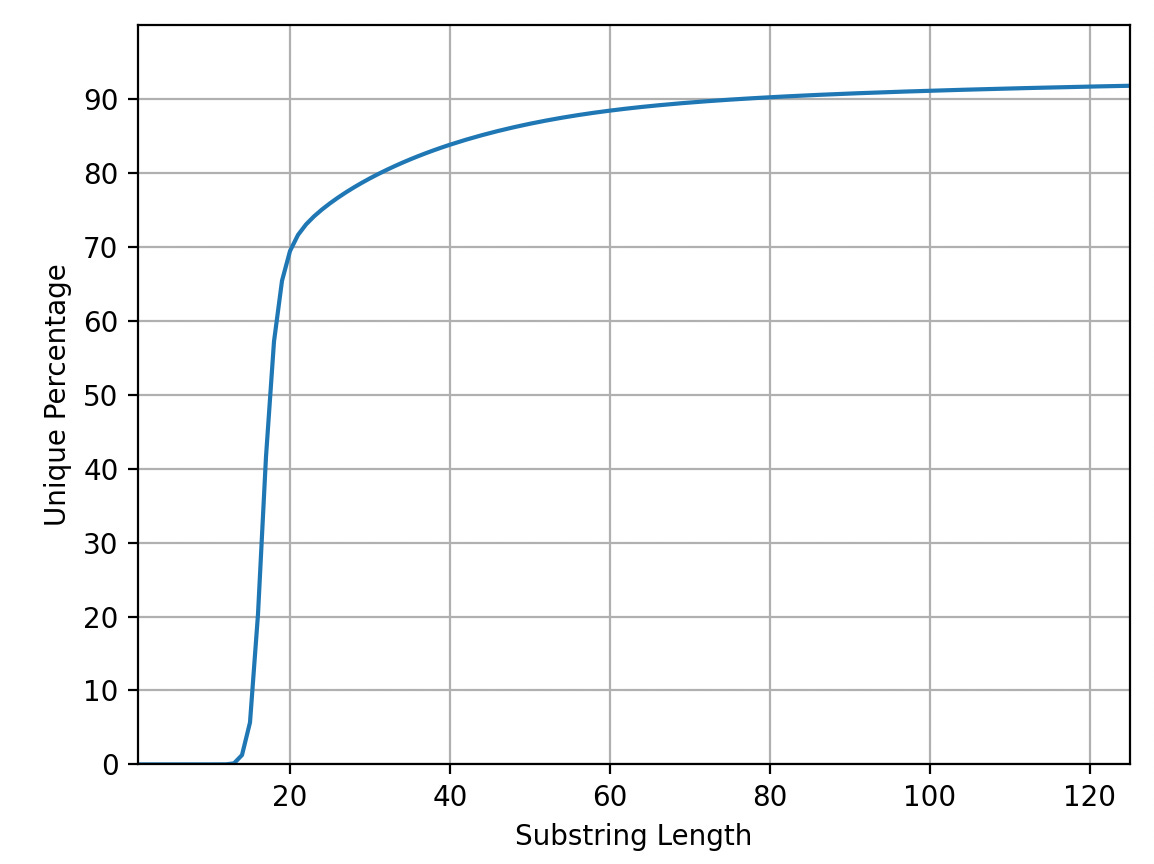
We now seem to be a little over 70% at 25bp so things have dropped by ~10%. However I we’re still addressing the majority of coding regions. If we look at the final 5% it seems like there are diminishing returns after ~20Kb reads:
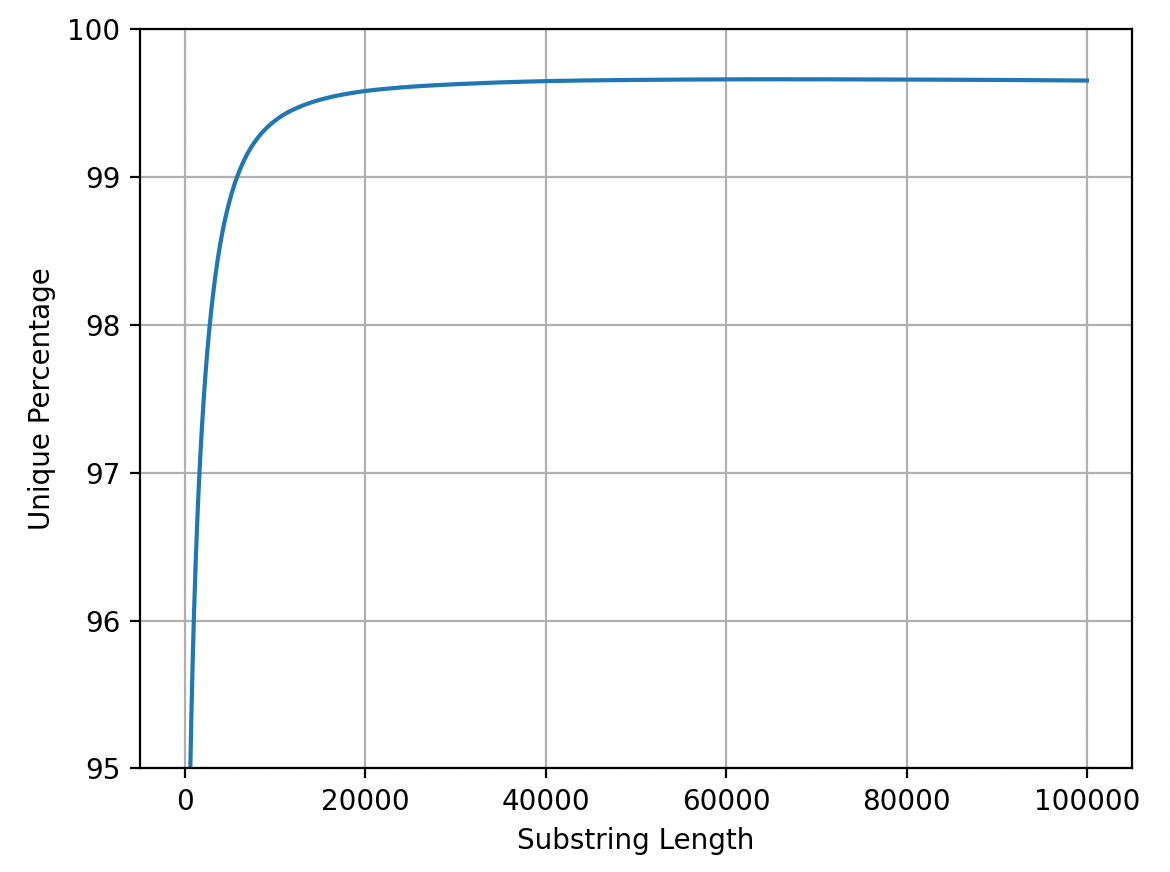
Intuitively this makes sense as I’d imagine the vast majority of repeats are going to be much shorter than 20Kb… In any case long reads give us a way to address 5 to 10%9 of the genome which we could never unambiguously address with short reads alone.
Would you like to see this analysis for the forward and reverse strand? Just one annual subscription could buy this suffering Computer Scientist a 256Gb memory upgrade… or enough AWS credits for a week.
Anyway those are the more serious conclusions of this analysis. Now let’s look at some more colorful plots!
One of the other pieces of analysis this code enables is what we called the “repeat score plot”. It’s basically a way of looking at “the number of things that repeat X times”:
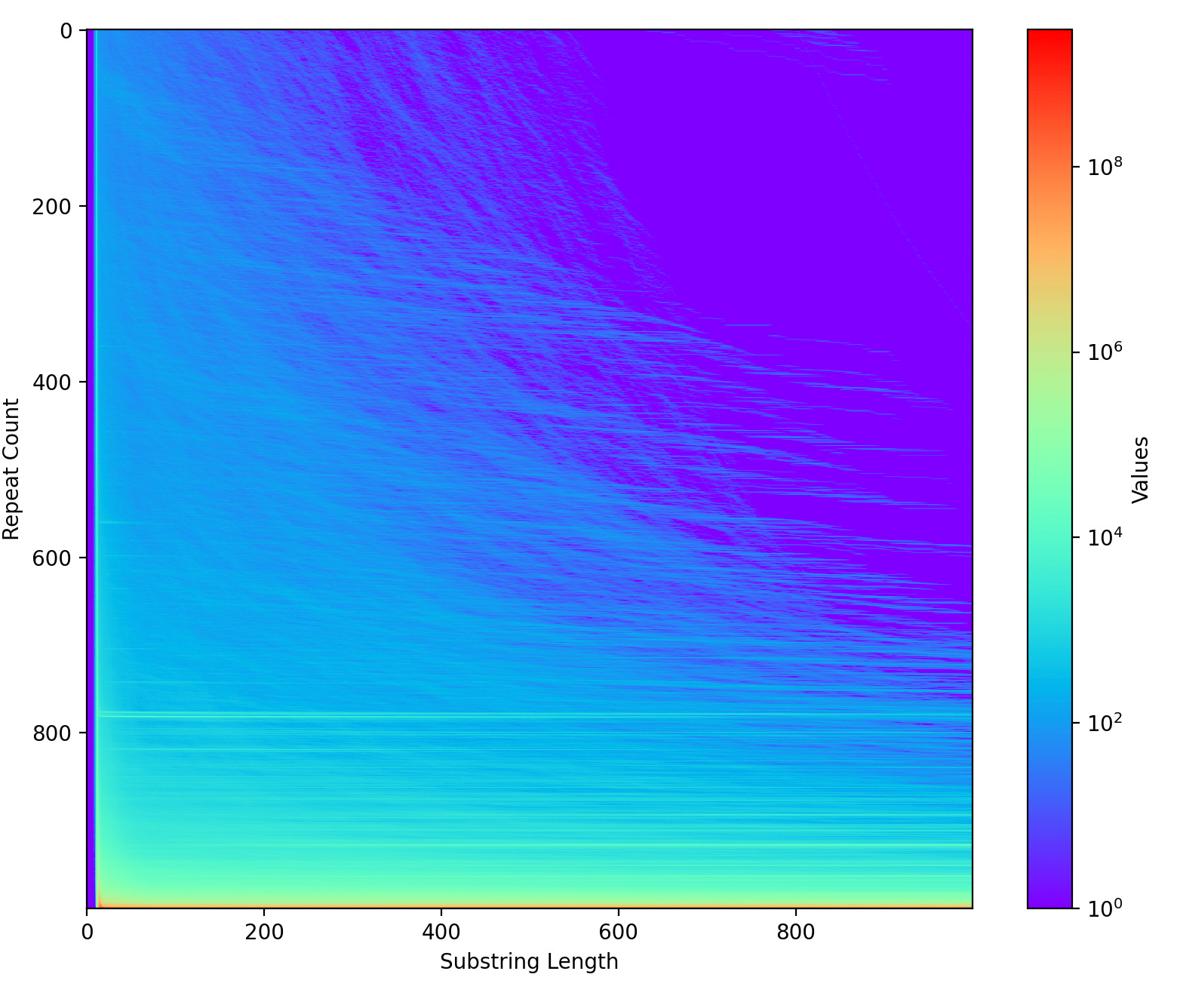
The horizontal lines at ~800 in the plot above suggest some exact duplicates of >1000bp that repeat 800 times. The “furry looking” stuff in the top half of the plot? These will be inexact repeats. As copies diverge from each other the number of repeats decreases creating these wispy furry things:
Mostly this is just a fun piece of analysis…
But the uniqueness calculations and general understanding of the repeat structure feels like a nice analysis to have in your back pocket when evaluating new approaches.
It would also be fun to dig out some of the longer repeats and have a list of “difficult” regions. Classifying the large number of exact or near exact repeats >300bp could be interesting. The work we previously did suggested the majority of these repeats were in non-coding regions…
We also tried running non-genomic sequences. Here dialogue and stage directions in Shakespeare:

Somewhat predictably, the stage directions were much more repetitive. Stretching an analogy we can suggest that if “stuff” repeats, perhaps one possibility is that it has some kind of coordinative function…
In any case as always, subscribe for more of this nonsense!
That is given the human genome reference we had available at the time.
They must have been a pretty switched on investor to have seen our paper to be honest!
In this very limited context.
I wanted to be able to run it efficiently over all substring lengths so developed a suffix array based tool that would do this efficiently. Even so, the tool required 60Gb+ of RAM. Pretty much everything was 32bit at the time and limited to 4Gb… The university had one weird server with ~80Gb of RAM… it was some weird hybrid system… 32bit with 64bit addressing. Even so… the code had to be written to use 33bit addressing as full 64bit addresses would have doubled the sizes of the data structures.
I actually ran this analysis on the old HiSeq X server I previously mentioned.
At some point I pulled out the 33bit array so everything is now using 64bit addressing. I could probably revert this… but I don’t really have the time or the inclination at the moment.
GCF_009914755.1_T2T-CHM13v2.0_genomic
There seems to be at least one error compensating for breaks between contigs/chromosomes. This should significantly alter the results. But for a substack post I can’t justify looking deeply into this.
The plot below shows the 90 to 100% region. You get above 95% at ~500bp reads.

Very interesting, thanks Nava. Some thoughts:
1) Any idea what's the 0.5% of the genome that can't be resolved even at 100k readlength (from your 3rd graph).
2) Fig 4 and 5 have opposite Y axes; is Fig 4 Y axis inverted?