


The PacBio $500 Genome And SPRQ
PacBio announced a new chemistry for the Revio called SPRQ today. At a high level this is what it enables:
120Gb per Revio chip (up from 90Gb) - enabling $500 20x Human Genome
Instrument cost has dropped to $599,000
Lower input requirements (down to 500ng).
As I understand it that 120Gb is the baseline specification for the kit. I had a chance to chat with the PacBio folks today and they showed one run with:
134.7Gb composed of 8.7M HiFi reads, Q35 median read quality, 15.55Kb mean read length.
This seems very compelling. A $500 20x Q30 human genome doesn’t seem like a difficult sell over a $200 Illumina genome. Or a lower baseline accuracy Nanopore genome of indeterminate cost1.
I was of course interested in what exactly has let them boost run throughput. Previously I’d put together this estimate of read distribution:
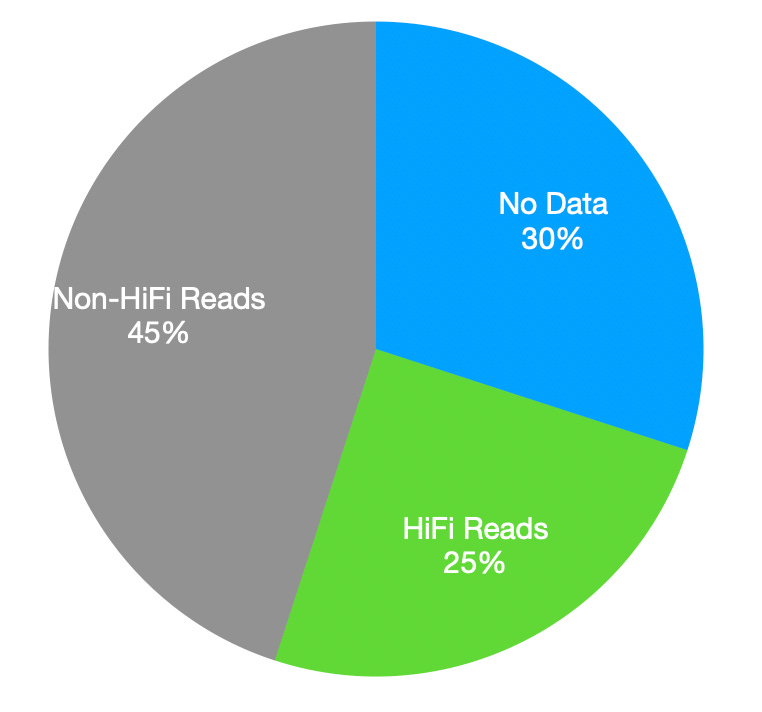
They said it was a combination of factors. Firstly the chemistry changes they’ve made to improve loading2 (and let them get down to 500ng input) appear to allow them to load previously inaccessible parts of the chip.
You can see some variation across the chip in the heat maps I previously generated. But I’m not sure this would explain a 30% boost in throughput. They also stated that they’d done significant polymerase engineering, and this had a somewhat larger contribution to throughput improvements3.
The mean number of “HiFi passes” was 9 in this new dataset. Older Revio datasets also showed an average of 9 passes. So I suspect filters remain unchanged. It’s just that on average the polymerase lives longer, and the filter now lets through a larger fraction of the data.
If we assume that the“No Data” segment of the pie is unchanged, but the “Non-Hifi” piece has got smaller. This would suggest the following breakdown for the new chemistry:
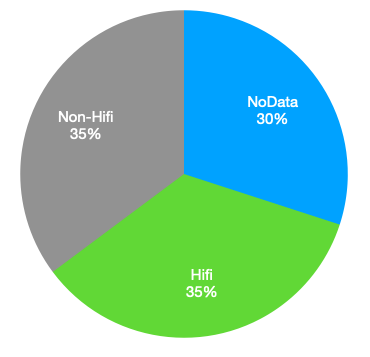
This is a solid improvement, and quite compelling. But it also seems like there’s still a ton of potential for higher throughput runs on the Revio through further chemistry improvements.
In terms of how this might impact the sequencing market, that’s another post for another day. Stay subscribed!
Given Oxford Nanopore still don’t seem to provide any kind of throughput spec for their instruments a cost is difficult to calculate, but perhaps I’ll give it a go in a future post. I’m also basically of the opinion that PacBio data is higher quality and less biased than Oxford Nanopore (but PacBio clearly can not provide as long reads). The 15 to 20Kb range however seems more than sufficient for human genomics (opinions may vary).
PacBio provided the following note on loading improvements:
We're basically using the sample more effiently by increasing the local concentration near the chip surface. ZMW loading is mainly driven by pseudo 2D diffusion of complexes on the surface; this lets us push more DNA onto the surface. You could think of the surface having regional variations in binding capacity and permitivity to diffusion as well…
…with a higher solution concentration we're pushing that surface diffusion harder and doing better with less cooperative regions. Still, the main contributor to higher yields is a decrease in early terminations, bringing up the fraction that make it into the HiFi category. That alone would tend to decrease mean n passes, but we're doing somewhat better on overall survival too, pushing back the other way. As to future potential, we're probably still under 50% or less of TMO, but a lot of that headroom is difficult to get to without true super-poisson loading.
PacBio also noted that this was all through chemistry changes, and there are no chip modifications here!
I think a safe $ estimate for a 20-25x Promethion run is around $700. Of course, theoretically it can do more (sometimes over 30x) but then you start running into issues of flow cell variability and the moment you need to use two flowcells it jumps to $1400.
This is only half the issue with human implementations though. Errors made by the ONT systems are not as well accounted for in the variant calling pipelines, especially compared to Illumina's. This is a general statement with many nuances but the inevitable outcome is the following: You will get pathogenic variants you must validate at much higher rates, and the cost of these variants will send your per patient costs to the moon. Furthermore, the majority of these will come back false positive, as was my experience. In other words, if the variant calling isn't as accurate, this costs from this will be a comparable or even larger pain point. I'd love to see more data on that whole implementation from PacBio.